
HEMOCROMATOSE
A hemocromatose hereditária (HH) é a mais comum doença genética, hereditária, na população caucasiana (branca), alcançando até 1 em 200 pessoas descendentes de nórdicos ou celtas. Trata-se de uma predisposição para a absorção excessiva de ferro da alimentação. Este ferro acumula-se principalmente no fígado, pâncreas e coração, podendo levar ao óbito por cirrose, hepatocarcinoma, insuficiência cardíaca ou diabetes. No quadro abaixo iremos ver a classificação das hemocromatoses



| CLASSIFICAÇÃO DAS HEMOCROMATOSES | | | |
| Hemocromatose Hereditária | associada ao HFE (tipo 1) | C282Y homozigose | |
| C282Y/H63D heterozigose composta | | ||
| não associada ao HFE | hemocromatose juvenil (tipo 2) | mutação hepcidina (2A) | |
| mutação hemojuvelina (2B) | |||
| hemocromatose tipo 3 (mutação do receptor 2 da transferrina) | | ||
| mutação da ferroportina tipo 4 (hemocromatose autossômica dominante) | | ||
| Sobrecarga de ferro adquirida (secundária) | anemias carregadoras de ferro | | |
| talassemia maior | | | |
| anemia sideroblástica | | | |
| anemia hemolítica crônica | | | |
| sobrecarga dietética de ferro (Africana) | | | |
| sobrecarga parenteral de ferro (incluindo politransfusão) | | | |
| Outras causas (raramente no grau observado na HH) | hemodiálise prolongada | | |
| hepatopatia crônica | | | |
| | | ||
| | | ||
| | | ||
| porfiria cutânea tarda | | | |
| síndrome da sobrecarga de ferro dismetabólica | | | |
| pós shunt portocava | | | |
| sobrecarga de ferro na Africa sub-sahariana | | | |
| sobrecarga de ferro neonatal | | | |
| aceruloplasminemia | | | |
| atransferrinemia congênita | | |
Sintomas
Uns dos sintomas na maioria dos casos são:
Fadiga
Fraqueza
Dor abdominal
Perda de peso
Amenorréia (ausência de menstruações)
Dor nas juntas
Insuficiência hepática (fibrose, cirrose, etc.)
Carcinoma hepatocelular (câncer de fígado)
Diabetes
Insuficiência e arritmia cardíacas.
Diagnóstico
A recomendação da AASLD (Associação Americana para o Estudo de Doenças do Fígado) para o diagnóstico da hemocromatose hereditária é um algoritmo que leva em consideração as características e o alto custo dos exames (o estudo genético custa cerca de US$ 2.700) para detectar o maior numero possível de doentes. Este algoritmo de 3 passos começa com a detecção daqueles com acumulo de ferro, depois com a confirmação genética e posteriormente avalia o grau de lesão de órgãos.
| | |
| Sintomáticos | Manifestações inexplicáveis de doença hepática ou com alteração dos exames indiretos de ferro |
| | Diabetes mellitus tipo 2 com hepatomegalia, alteração de enzimas hepáticas, doença cardíaca ou disfunção sexual precoce |
| | Doença articular atípica precoce, cardiopatia e disfunção sexual masculina |
| Assintomáticos | Parentes de primeiro grau de indivíduos com hemocromatose |
| | Alteração dos exames indiretos de ferro em exames de rotina |
| | Elevação de enzimas hepáticas, hepatomegalia ou aumento da atenuação do fígado em tomografia |
Etiologia
Na hemocromatose hereditária, mutações genéticas (geralmente transmitidas de pais para filhos) levam a um aumento na absorção do ferro no intestino (duodeno e jejuno proximal), o que leva ao acúmulo do metal no organismo.
Desde a descoberta do gene HFE, que regula a absorção de ferro, duas mutações nesse gene foram descritas e são responsáveis pela maioria dos casos de hemocromatose hereditária: a C282Y e a H63D. Famílias com hemocromatose sem ambas as mutações, no entanto, mostram que outros genes devem estar envolvidos.
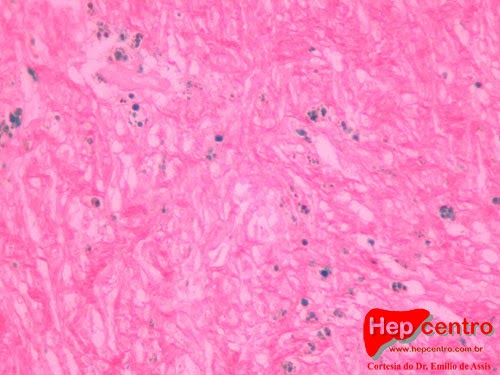
Biopsias hepáticas com hemocromatose. Na primeira, corada com HE, observa-se o ferro como granulações alaranjadas. Na segunda, corada com Perls, o ferro aparece azul.
Evolução Clinica
A doença inicia-se com o acumulo lento e insignificante de ferro no organismo (0-5 g). Em um segundo estagio, já ha excesso de ferro (10-20 g) que, se não tratado, pode levar a lesão de órgãos, geralmente apos 40 anos de idade e mais que 20 g de ferro acumulado. Nosso objetivo, portanto, é detectar estes pacientes antes da terceira fase. Felizmente,marcadores indiretos do ferro acumulado permitem o diagnostico precocemente e prevenir o aparecimento da doença.
Tratamento
O tratamento utilizado globalmente, de eficácia comprovada, relativamente barato e praticamente inócuo é a flebotomia (sangria) periódica, ou seja, a retirada de sangue. As células vermelhas do sangue contem hemoglobina, da qual o ferro é componente importante (normalmente, 70% do ferro no nosso organismo está concentrado na hemoglobina). Com a retirada periódica de sangue (nos pacientes com ferritina sérica > 1.000 ng/ml, geralmente uma bolsa de sangue por semana, com nova dosagem de ferritina a cada 4 a 8 semanas), ocorre o deslocamento do ferro depositado nos tecidos para a formação de novas moléculas de hemoglobina, até que não há mais excesso (no início do tratamento, definido como ferritina sérica menor ou igual a 20 ng/ml). Depois, são realizadas novas flebotomias em intervalos variáveis procurando manter o valor de ferritina sérica menor ou igual a 50 ng/ml. Com esses cuidados, não costuma surgir anemia significativa durante o tratamento e a evolução da doença é interrompida, não levando a progressão das lesões nos órgãos-alvo.
Molécula de hemoglobina. A porção verde é a fração "heme", que contém ferro.
Outra opção de tratamento, usada especialmente nos que não toleram a flebotomia pela presença de anemia, é a quelação (uso de medicação que se une a íons metálicos e é eliminada do organismo). O único agente quelante utilizado disponível até há pouco tempo era a desferrioxamina (DFO), medicação usada por via endovenosa geralmente com uma bomba de infusão, pois a aplicação deve ser feita lentamente (entre 8 a 12 horas) ou por via subcutânea. Apesar dos benefícios, a medicação é tóxica, levando a irritação no local da aplicação, deformidades ósseas, retardo no crescimento de crianças e efeitos neurotóxicos visuais e auditivos.
Depósitos (azuis) de ferro na biópsia hepática
Recentemente, a deferiprona e o deferasirox foram introduzidos como opções mais acessíveis, pela sua administração oral, eficácia comparável à desferrioxamina e menor índice de efeitos colaterais. No entanto, pode levar a efeitos colaterais como agranulocitose, leucopenia, artralgia, náuseas e vômitos, dor abdominal e hepatotoxicidade, não sendo, portanto uma opção razoável à flebotomia nos pacientes sem contra-indicação a esse procedimento.
SURDEZ
É enorme o número conhecido de doenças genéticas heterogêneas, ou seja, que exibem fenótipos muito semelhantes, porém são condicionadas por mecanismos genéticos diferentes, por genes situados em locos distintos (heterogeneidade não-alélica) ou ainda por alelos diferentes, em diversas combinações, de um mesmo gene (heterogeneidade alélica). Com os avanços da biologia molecular, a cada dia são descobertos novos genes causadores de doenças tidas, até então, como determinadas por um único loco ou alelo. O aconselhamento genético e do diagnóstico clínico de anomalias hereditárias, é importante que se conheça e se leve em conta o grau de heterogeneidade dessas doenças, pois além de esse fenômeno ser causa importante de variação clínica, é fundamental para a correta estimativa dos riscos de recorrência.
SOBRE SURDEZ CONGÊNITA
A surdez é um defeito cuja incidência é significativa na população, podendo ser considerada um problema importante de saúde pública. Ela assume uma importância considerável dentro das deficiências congênitas ou adquiridas e traz graves conseqüências para o desenvolvimento da criança e da sociedade. A surdez congênita é uma afecção comum que está presente nas crianças em freqüência que varia, segundo os vários autores, de 1 : 2000 a 1 : 600, dependendo da região ou país. A incidência do defeito varia nas diferentes populações como conseqüência de variação de fatores ambientais, capacidade diagnóstica e cuidado com a saúde local; nos países desenvolvidos, cerca de uma em cada 1000 crianças nasce com alguma deficiência auditiva significante e mais ou menos metade (1/2000) apresenta surdez hereditária. Segundo dados da Organização Mundial de Saúde (OMS), 10% da população mundial apresenta algum tipo de problema auditivo devendo, portanto, existir cerca de 15 milhões de deficientes auditivos no Brasil, sendo 350 mil indivíduos portadores de surdez profunda. Pelo Censo Demográfico de 1991 (Fundação Instituto de Geografia e Estatística - IBGE), existem cerca de 174.000 casos de deficiência auditiva no Brasil (0,12% da população). Estima-se que pelo menos 1,13% da população brasileira apresente algum tipo de deficiência; entre os afetados, 10,41% são surdos. Nos países desenvolvidos tem ocorrido nas últimas décadas uma diminuição acentuada das causas ambientais de doenças, o que, consequentemente, tem aumentado a importância relativa das doenças genéticas e parcialmente genéticas causadoras da morbidade e mortalidade, principalmente em crianças. Já aqui no Brasil, como existe entre nós, em relação à surdez congênita, um excesso de casos ambientais, originários de infecções ocorridas durante a gravidez e de má assistência médica durante a gestação, parto e período neo-natal, espera-se que o risco de repetição seja diferente do encontrado nos países do primeiro mundo, cujas estatísticas são usadas tradicionalmente para se compor os riscos de recorrência levando-se em conta a proporção de casos ambientais e genéticos. . A prevenção da surdez deverá auxiliar na redução desse custo destinado à educação especial da criança surda e do seu tratamento médico.
ETIOLOGIA DA SURDEZ
Nas pesquisas internacionais com relação à surdez congênita indica que 46 a 60% de todos os casos de disacusia são geneticamente determinados. Em 70% dos casos, a surdez é não sindrômica (isolada), podendo apresentar qualquer um dos tipos de herança monogênica.
As freqüências dos diversos tipos do defeito são (esses valores variam de acordo com a região e com o país):
- hereditária - mecanismo autossômico recessivo: 60%-75%; mecanismo autossômico dominante: 20%-30%; mecanismo recessivo ligado ao X: 2%-3%;
- ambiental ou fenocópia: 20%.
Normalmente a surdez genética é congênita, mas algumas formas surgem e progridem na idade adulta (surdez dependente da idade) até a completa perda da audição; esse fenômeno é comum quando a surdez é autossômica dominate. Fatores genéticos são responsáveis pela suscetibilidade à surdez, mesmo não-genética, de início tardio de algumas pessoas. Outras formas dependem inteiramente de fatores ambientais para se manifestarem.
Muitos casos de surdez, principalmente em países em desenvolvimento como o nosso, são ambientais. Muitas vezes, é difícil a distinção entre surdez geneticamente determinada e surdez congênita adquirida. Perdas auditivas congênitas, desencadeadas durante o período pré-natal (fetal) devido a infecções e outras causas.
| Fatores ambientais causadores de disacusia, em algum estágio do desenvolvimento infantil | ||
| Pré-natal | Infecção materna (rubéola, citomegalovírus, toxoplasmose, sífilis, etc) | |
| Terapia materna com drogas ototóxicas | ||
| Irradiação (raios-X) durante gestação | ||
| Peri-natal | Icterícia | |
| Trauma de parto | ||
| Anoxia | ||
| Pós-natal | Otites | |
| Meningite | ||
| Sarampo | ||
| Caxumba | ||
| Traumatismos | ||
| Drogas (antibióticos aminoglicosídicos) | ||
SURDEZ DE ETIOLOGIA GENÉTICA
Muitos genes contribuem para o desenvolvimento normal da audição humana. Consequentemente, defeitos em qualquer um desses genes poderão levar à deficiência auditiva. Já foram descritas cerca de 60 tipos de deficiência auditiva genética no homem que podem ser razoavel ou parcialmente distinguidas umas das outras com base no padrão de herança, freqüência, idade de início, severidade e local de lesão. A maior parte dos casos de surdez hereditária é devida a genes recessivos que se expressam no epitélio sensorial do ouvido interno.
| Número de locos envolvidos na surdez | |
| Tipo de surdez | Número de locos |
| Autossômica recessiva | 20-45 |
| Autossômica dominante | 15 |
| Ligada ao X recessiva | 8 |
| Surdez hereditária | 73 |
SURDEZ RELACIONADA A SÍNDROMES GENÉTICAS
A surdez é uma característica freqüente em mais de 100 síndromes hereditárias ou genéticas conhecidas. A tabela abaixo mostra algumas dessas síndromes, nas quais a disacusia é considerada uma característica relevante.
| Exemplos de síndromes que apresentam deficiência auditiva. | ||
| Surdez com malformações craniofaciais e cervicais | Síndrome de Treacher Collins | |
| Síndrome de Crouzon | ||
| Síndrome de Apert | ||
| Surdez com displasias esqueléticas | Osteogênese imperfeita | |
| Síndrome de Klippel-Feil | ||
| Surdez com anomalias de derme | Síndrome de Waardenburg | |
| Surdez com disfunção renal | Síndrome de Alport | |
| Surdez com anomalias oculares | Síndrome de Usher | |
| Síndrome de Fraser | ||
| Surdez com disfunção metabólica | Síndrome de Pendred | |
| Síndrome de Hurler | ||
| Síndrome de Hunter | ||
| Surdez com anomalias cromossômicos | Trissomia do cromossomo 18 | |
| Trissomia do cromossomo 21 | ||
PADRÕES COMPLEXOS DE SEGREGAÇÃO NA SURDEZ GENÉTICA
Vários padrões complexos de segregação são possíveis devido a casamentos preferenciais entre afetados que vivem em agregados ou em comunidades próprias. Em vista de uma precoce segregação educacional e social e em vista das dificuldades de comunicação com ouvintes normais, essa tendência a casamentos preferenciais é completamente natural. Por exemplo, um indivíduo com surdez dominante pode casar-se com uma pessoa portadora de surdez recessiva. A prole afetada desse casamento apresentará surdez autossômica dominante, mas será também heterozigota para o alelo causador de surdez autossômica recessiva. Se alguma dessas pessoas se casar com alguém portador do mesmo tipo de surdez autossômica recessiva, então tanto a surdez recessiva como a dominante podem ocorrer entre seus descendentes. Esse tipo de segregação complexa ocorre na população e pode ser extremamente difícil de ser detectada na prática.
Postado por : Sarah Ferreira Couto
Referências
- Bret L. Hicken, Diane C. Tucker, James C. Barton. Patient compliance with phlebotomy therapy for iron overload associated with hemochromatosis. The American Journal of Gastroenterology, Volume 98, Number 9 (September 2003), pp. 2072-2077
- Barton JC, McDonnell SM, Adams PC, et al. Hemochromatosis Management Working Group: Management of hemochromatosis. Ann Intern Med 1998; 129:932–9.
- Fabron JR, Antonio and Tricta, Fernando. Oral iron chelator therapy with deferiprone in patients with overloaded iron. Rev. Bras. Hematol. Hemoter., 2003, vol.25, no.3, p.177-188
Zeid Kayali, Rostislav Ranguelov, Frank Mitros, Chrisandra Shufelt, Farshad Elmi, Stephen C. Rayhill, Warren N. Schmidt, Kyle E. Brown. Clinical Studies Hemosiderosis is associated with accelerated decompensation and decreased survival in patients with cirrhosis . Liver International, Volume 25, Number 1 (February 2005), pp. 41-48
Eugenia Lauret, Manuel Rodríguez, Segundo González, Antonio Linares, Antonio López-Vázquez, Jes Martínez-Borra, Luis Rodrigo, Carlos López-Larrea. HFE gene mutations in alcoholic and virus-related cirrhotic patients with hepatocellular carcinoma. The American Journal of Gastroenterology, Volume 97, Number 4 (April 2002), pp. 1016-1021
Nenhum comentário:
Postar um comentário